SnapGene Version 5.0.0
SnapGene 5.0.0 was released on September 22, 2019.
Overview
Version 5.0 adds new capabilities and display options including pairwise alignment, import from the Ensembl database, support for directional TOPO® cloning, and improved tools for alignment to a reference DNA sequence.
Pairwise Alignment
Pairs of DNA or protein sequences can be analyzed by local, global, or semi-global alignment.

Import from Ensembl
Gene or transcript data from the Ensembl genome browser can now be imported directly into SnapGene.

Directional TOPO® Cloning
A new interface simulates directional TOPO® cloning into topoisomerase-activated vectors.

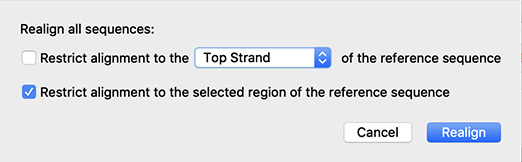
Flexible Alignment to a Reference
The interface for aligning to a reference DNA sequence has been enhanced. Controls for various display options are more intuitive, and alignment can be restricted to a designated strand or region of the reference sequence.
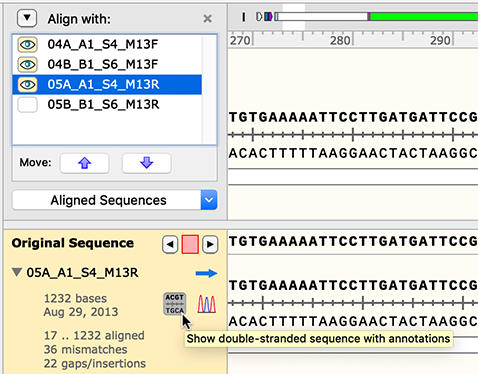
Draggable Nonaligned Ends
For a sequence that has been partially aligned to a reference DNA sequence, the nonaligned end portions can now be dragged out and visualized.

Customizable Background Color
The background color for the SnapGene interface can be changed.

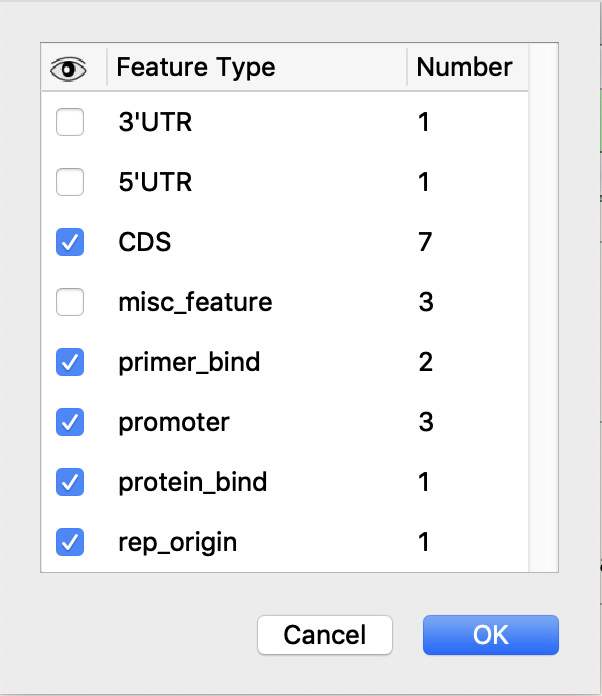
Visibility Controls for Feature Types
Features can be shown or hidden based on feature type.
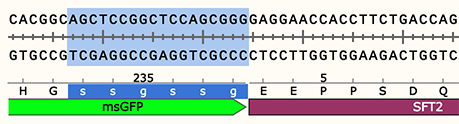
New Options for Amino Acids
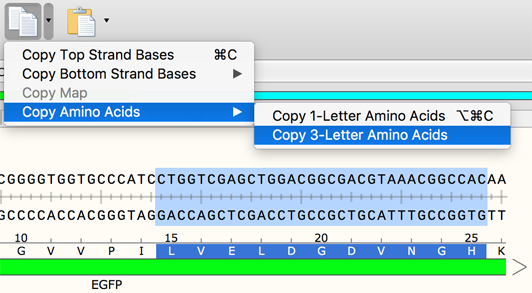
Amino acids in a translated feature can be set to lowercase, and a stretch of amino acids can be copied in 3-letter format.
Codon Frequencies Calculator
A codon frequencies table can be generated for one or more translated features.

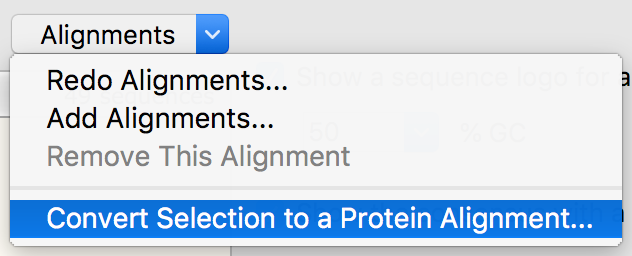
DNA-to-Protein Alignment Conversion
A selected region of a DNA alignment can be translated to generate the corresponding protein alignment.
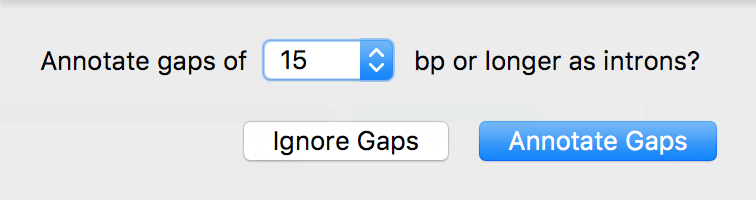
Intron Annotation for an Aligned cDNA
When a cDNA is aligned to a reference genomic DNA sequence, the cDNA can be used to create a feature in which gaps are annotated as introns.
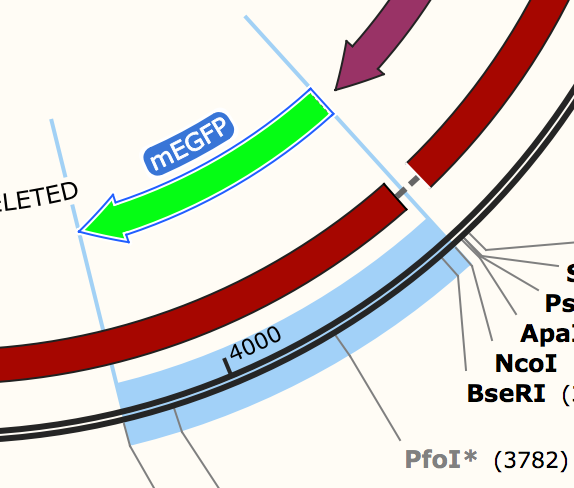
Extended Selection Endpoints in Maps
Selections are now marked with extended lines, which clarify the selection endpoints and also enhance visibility for small selections.
Importer for DNASIS Files
Files from the legacy DNASIS program are now recognized by SnapGene.
New Functionality
- Added tools for pairwise alignment of DNA and protein sequences.
(Requested by many customers) - Added interfaces for simulating directional and blunt TOPO® cloning.
(Requested by many customers) - Provided support for customizing the background window color.
- Added the ability to drag out and view non-aligned ends of aligned sequences.
(Requested by just about everyone) - Added the ability to display feature translations in lowercase.
(Requested by Peter Drain) - Enabled alignments to be constrained to a designated strand or region of the reference DNA sequence.
(Requested by Christel Aebischer and others) - Added the ability to import sequences directly from Ensembl.
(Requested by Jaeho Ryu, Elena Fujiwara, and others) - Enabled the calculation and saving of codon frequencies for one or more translated features.
(Requested by Craig VanDolleweerd and Jeanie Lin) - Enabled feature visibility to be toggled by feature type.
(Requested by Di Xia and Lee-Chung Lin) - Added the option to trim low-quality ends when importing sequence traces for contig assembly or multiple sequence alignment.
- Enabled an aligned cDNA to be used to annotate a feature with exons and introns.
(Requested by Rob Steele and Peishan Yi) - Added support for opening DNASIS files.
(Requested emotionally by Jim Galen) - Enhanced BLAST support to provide all five options (blastn, blastx, tblastx, blastp, and tblastn).
(Requested by Inbar Plaschkes) - Updated the "Aligned Sequences" menu when viewing an alignment to a reference sequence, and added the following new commands: • Duplicate Selected Sequence(s) in New Window(s) • Remove All Sequences • Show/Hide Quality Data for Sequence Traces
- Added a control in Preferences to enable quality data for sequence traces to be shown by default when aligning to a reference DNA sequence.
- Enabled a selected portion of a multiple DNA alignment to be converted to a multiple protein alignment.
(Requested by Walid Azar) - Added a control for exporting selected files in a collection as a list in PDF or tab-separated format.
(Requested by David Murray) - Added a "Choose DNA Sequences..." button to the Simulate Agarose Gel dialog, as a shortcut for configuring multiple lanes.
(Requested by Dhaval Bhatt) - Enabled amino acid sequences to be copied as either 1- or 3-letter amino acid codes.
(Requested by Moo-Hyung Lee) - Enabled the copying of selections that span multiple lines in pairwise or multiple alignment windows.
- Added the New England Biolabs "TriDye™ Ultra Low Range DNA Ladder".
Enhancements
- Calculated the total length of the segments for a feature with gaps, with this information now displayed in Features view, feature dialogs, and feature tooltips.
(Requested by Di Xia) - Reduced the width of the "Code Number" column in the collection interface by using an icon.
(Requested by Carles Alvarez) - Added the option to preserve a feature translation by adjusting its reading frame when exiting the Edit Feature dialog.
(Inspired by Wulf-Dirk Leuschner) - Added an option in Preferences to display the MW of a selection in a protein file in Daltons.
(Requested by Eric Fang) - Added a notification if some of the input sequences were not included when assembling contigs.
(Requested by CLS) - Added a keyboard shortcut for importing primers from a list.
(Requested by Benjamin Weinberg) - Relaxed a restriction that prohibited < and > characters in primer names.
(Requested by Tomoyuki Kosaka) - Improved the detection of T7 promoter features.
(Reported by Kevin McGowan) - Improved import from NCBI to tolerate regions in which the left and right endpoints are swapped, and to recognize "c" as denoting the reverse complement.
(Requested by Rachel Delston) - Added support for new genetic codes.
(Requested by Dilbag Multani) - Improved the algorithm for aligning to a reference DNA sequence.
(Requested by Leonid V.) - Improved the import of features from BED and GTF files generated by geneXplain.
(Requested by John Bass) - Enhanced the Vector NTI® database importer to recognize separators.
(Requested by Sanofi) - Provided the version numbers of alignment programs (e.g., MUSCLE) in the user interface.
- Improved the visibility of selections and their endpoints in maps by including lines that extend below the DNA line.
- Enhanced the "Edit MW Markers List" dialog to support display of MW markers by supplier, and to enable one-step selection of all MW markers from a supplier.
- Dramatically improved scrolling performance in Features view for sequences with many features.
- Added commands to the Enzymes and Primers menus for pulling up the corresponding tabs in Preferences.
- Enhanced Properties view in protein windows to list both average and monoisotopic molecular weights.
- Made the alignment dialogs modeless to enable switching to other SnapGene windows.
- Improved the visibility of a Consensus selection in a multiple alignment window by extending blue lines down from each end of the selection.
- Made extensive optimizations to speed up application launch and use in general.
- Switched from WebKit to QTextEdit for rich text controls.
- Improved the support for links in feature qualifiers and in Description Panel fields.
- Enhanced the TA and GC Cloning dialogs to allow simple issues (e.g., lack of 5' terminal phosphates or overhangs) to be addressed by clicking a button.
- Improved the controls for toggling the visibility of sequences aligned to a reference sequence.
- Improved the controls for switching between showing an aligned sequence trace as double-stranded DNA or a chromatogram.
- Added a warning that only Clustal Omega can preserve internal stop codons when computing a multiple sequence alignment.
- Enabled a Main Collection to be specified if necessary when using "Save to Main Collection".
- Added "List View" and "Folder View" commands to the View menu when browsing a collection.
- Moved the "Design Synthetic Construct" and "Order Construct" commands to the Tools menu.
- Consolidated the default font size settings for all file types (DNA, protein, alignments), and added support for the default font size for alignments.
- Improved the discoverability of using Opt/Alt to see all four base heights when mousing over a peak in a sequence trace.
- Added a "Manage Codon Usage Tables" command to the Tools menu, because previously this option was only accessible from the "Codon Usage Tables" dialog.
- Ensured that rich formatting (bold, italic, underline, super- and subscripts) are visible in Features view even when full descriptions are not shown.
- Added "GC Content" values to the DNA Calculations window.
- Enabled export of gel images, maps, and histories to the EMF vector format on macOS for import into Microsoft Office products.
- Added "Show/Hide All Features" commands to the Features menu.
- Added "Show/Hide All Primers" commands to the Primers menu.
- Ensured that when importing from NCBI, the LOCUS field is used as the default file name.
- Enhanced the logic so that when importing from the SnapGene online database, a perfect match to the query is automatically selected even if it is not the first match listed alphabetically.
- Added the ability to transfer a selection between the Consensus and an aligned sequence in a multiple alignment window by using Cmd/Alt + click.
- Added keyboard shortcuts for the "Flip Sequence" and "Set Origin" commands.
(Requested by Michael Lemieux) - Enhanced the flexibility of a protein search to strip a terminal stop codon from the query if appropriate.
- Improved support for copying maps, history, and agarose gels and pasting in vector format into Microsoft Office on macOS.
- Added support for the following qualifiers with the following feature types. exon & intron: /trans_spicing; misc_recomb: /recombination_class; rep_origin: /function; source: /metagenome_source; tRNA: /operon.
- Improved the options available for the feature qualifiers /recombination_class, /regulatory_class, /db_xref, /gap_type, and /ncRNA_class.
- Added a shortcut for "Set Translation Numbering".
- Improved the layout of enzymes in linear maps.
- Improved the genetic code choice for ORFs and non-feature translations when importing from NCBI and opening GenBank and EMBL files.
- Configured the tool for alignment to a reference DNA sequence so that flipping the reference sequence or resetting the numerical origin preserves the alignments instead of recomputing them.
- Improved the visibility of bands when using MilliporeSigma's DNA Molecular Weight Marker IV and VII.
- Added supplier information for the New England Biolabs BsaI-HF®v2 enzyme.
- Added supplier information for the Thermo Fisher (Invitrogen) Anza™ enzymes.
- Made various textual, color, and other visual enhancements.
Fixes
- Prevented copied gel images from being clipped when pasting into Microsoft Office.
(Reported by Christian Probst) - Fixed an issue with decoding some RTF-encoded content (primer lists, etc.).
(Reported by Elisabeth Boger) - Improved detection of T7 promoters and other non-translated features.
(Reported by Kevin McGowan) - Ensured that terminal phosphates are never shown at the end of protein sequences.
(Reported by Karl Brune) - Enabled closing of the License Agreement dialog by pressing the Escape key.
- Fixed the vanishing "Save As" dialog after importing a file and then choosing "Save" in the top toolbar Save button menu.
- Omitted the background color when printing or saving the EULA.
- Removed various irrelevant disabled menu commands when viewing protein sequence windows.
- Prevented the "Save" command in the top toolbar menu from changing to "Save All" when Opt is pressed.
- Corrected the translation of reverse directional features imported from BED and Genome Compiler files.
- Added a requirement that Insert sequences in the TA and GC cloning dialogs are < 50 kb.
- Corrected various standard plasmids to not include "(linearized)" in the map label.
- Corrected the endpoint modifications for various TOPO® vectors.
- Ensured proper display of super- and subscripts in feature qualifiers and Description Panel fields.
- Enabled the deletion of gaps in multiple sequence alignments.
- Improved stability with Undo after deleting gaps in multiple sequence alignments.
- Enabled "Edit → Delete" when a cursor is placed in an alignment window.
- Fixed various issues with menus updating to reflect the selection in a multiple sequence alignment after using the Select All and Select Range commands.
- Provided an "Edit → Delete Gap Characters" command when only gap characters are selected in an alignment.
- Disabled the Print and Save buttons in the collection interface top toolbar when no content is shown.
- Ensured that the proper position of cleavage arrows is maintained when setting the numerical origin.
- Improved the behavior of the controls in the Select Range dialog.
- Improved the accuracy of primer binding site calculations to ensure that long loops are not inappropriately favored over multiple hybridized stretches.
- Improved support for IP-based licenses, especially when a network connection changes or is slow or is briefly unavailable.
- Enhanced stability during Find operations.
- Prevented an issue with choosing a codon usage table that is incompatible with the currently selected genetic code in the Choose Alternative Codons dialog.
- Ensured that the Edit Codon Usage Table always shows the correct genetic code.
- Fixed an issue with using "Copy ORF Translation" from an ORF context menu when no amino acids are selected.
- Prevented hiding of the top toolbar when switching to Full Screen mode.
- Ensured the proper display of regions aligned downstream of a gap.
- Ensured that right-clicking does not display a context menu if the clicked window is not active.
- Corrected a regression in which recent searches were not listed in the Find control pull-down menus in Features and Primers views.
- Fixed an issue with pliancy and selection in Features view after toggling full descriptions.
- Enhanced stability when opening some CLC Bio files.
- Improved the highlighting of matches when cycling through search results in Features view.
- Improved the "Find" options in the Edit menu when using Enzymes, Features, and Primers views.
- Enabled Enter/Return and Shift-Enter/Return when using "Find ancestor" in History view.
- Improved the conversion of pasted rich content that contains unusual characters in the New DNA/Protein File dialogs.
- Ensured that the "Show/Hide Enzymes" command in the Enzymes menu is enabled only in Map and Sequence views.
- Removed unnecessary warnings about ORFs in Sequence view when displaying the sequence in compact mode.
- Prevented blank commands from being listed under Edit → Find when no items in a collection are selected.
- Corrected an issue with opening MSF files.
- Improved stability when using “Save All” while a collection is visible.
- Prevented blank menu actions under Edit → Find when neither a DNA nor a protein document is visible within a collection.
- Ensured that highlighted mismatches with the reference sequence are always shown in the right color.
- Improved the opening of MSF files.
- Prevented a hang that could occur when attempting to delete a restriction fragment containing a translated feature that spans the numerical origin.
- Corrected an issue that resulted in an empty file list initially being shown in a newly created collection after import from Ensembl.
- Improved stability when using File → Save All with many edited files in a collection.
- Fixed an issue in which previously computed information about alignment to a reference DNA sequence could be lost when an aligned sequence was unchecked in the list before saving the file.
- Disabled "Edit → Delete" when the cursor is placed while viewing a multiple alignment.
- Prevented an issue that sometimes could occur when restoring an original alignment to a reference DNA sequence.
- Fixed issues with undoing the realignment of sequences to a reference DNA sequence.
- Improved the behavior when replacing feature names in a collection.